Nukleosid-Analogon
Ein Nukleosid-Analogon (Mehrzahl Nukleosid-Analoga) ist ein Analogon eines Nukleosids, also eine synthetisch hergestellte Substanz, die einem natürlichen Nukleosid ähnelt. Nukleosid-Analoga werden als Medikamente zur Behandlung von Virus- und Krebserkrankungen eingesetzt und außerdem als Bausteine bei der Synthese modifizierter RNA. Nukleotid-Analoga unterscheiden von den Nukleosid-Analoga durch eine Phosphorylierung, Basenanaloga durch das Fehlen einer Pentose.
Chemischer Aufbau
Natürliche Nukleoside sind zusammen mit Phosphaten die Bausteine von DNA und RNA. Nukleoside setzen sich ihrerseits aus zwei Bausteinen zusammen, nämlich aus einer Nukleinbase und einem Zuckermolekül. Bei den Nukleinbasen handelt es sich um heterozyklische organische Verbindungen mit einem Purin- oder Pyrimidingrundgerüst, bei den Zuckermolekülen um eine Pentose, und zwar entweder um Ribose oder um Desoxyribose. Wenn die Ribose eines Nukleosids Ribose oder Desoxyribose mit einem, zwei oder drei Phosphatresten verbunden ist, spricht man von einem Nukleotid.
Wenn man Nukleoside modifizieren will, gibt es verschiedene Ansätze: Man kann die Verknüpfung zwischen dem Zucker und der Nukleinbase ändern, also den Zucker mit einem anderen Atom des Purin- oder Pyrimidin-Moleküls verbinden, man kann zusätzliche oder andere Atome bzw. Molekülgruppen in die Nukleinbase einbauen, oder man kann den Zucker verändern.
Je nachdem, ob im jeweiligen Nukleosid-Analgon ein Purin-Ringsystem oder ein Pyrimidin-Ring zu finden ist, unterscheidet man Purin- bzw. Pyrimidinanaloga.
Wirkungsmechanismen
Nukleosid-Analoga können in Zellen von Eukaryoten, Bakterien, Viren oder in vitro ganz unterschiedliche Effekte entfalten. Prinzipiell gehören sie zur Gruppe der Antimetaboliten, also zu einer Gruppe von Substanzen, die im Organismus wie natürlich vorkommende Substanzen von bestimmten Enzymen gebunden und zum Teil auch prozessiert werden. Beim Einsatz als Medikamente müssen sie von der Zelle aufgenommen werden und sind erst wirksam nach einer intrazellulären Phosphorylierung und damit einer Umwandlung in Nukleotid-Analoga. Nukleosid- bzw. Nukleotid-Analoga ähneln chemisch natürlichen Nukleosiden/Nukleotiden und können deshalb zum einen Enzyme blockieren, die Nukleinsäuren replizieren oder transkribieren und dadurch die Vermehrung von Zellen, Viren oder Bakterien stoppen. Zum andern können sie in Nukleinsäuren anstelle normaler Nukleoside/Nukleotide eingebaut werden, was zu Strangabbrüchen sowie zu einer Störung von Replikation, Transkription und DNA-Methylierung führen kann. Basenanaloga wie z. B. 5-Fluorouracil können zudem die Nukleotid-Synthese behindern und so den Nukleinsäurestoffwechsel beeinträchtigen. Einzelheiten des jeweiligen Wirkungsmechanismus sind bei den einzelnen Substanzen beschrieben.
Im Weiteren soll für „Nukleosid-Analogon“ und „Nukleotid-Analogon“ einheitlich der Begriff Nukleosid-Analogon verwendet werden.
Weiterhin können sie bei der Synthese von Nukleosid-modifizierter mRNA (modRNA), die beispielsweise bei der Entwicklung von Impfstoffen verwendet wird, benutzt werden. Dies führt zu einer Veränderung der Sekundärstruktur, was letztlich eine Erkennung von modRNA durch das angeborene Immunsystem mindert.
Beispiele
Azidothymidin
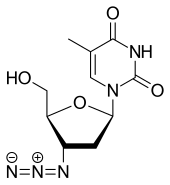
Azidothymidin (AZT, INN: Zidovudin) war der erste wirksame Arzneistoff gegen das HI-Virus. Da es am 3’-Kohlenstoff der Ribose statt einer Hydroxygruppe eine Azidogruppe aufweist, kann hier bei der Synthese der Virus-DNA die Kette nicht mehr weiterwachsen und es entsteht ein inaktives Provirus.
Puromycin
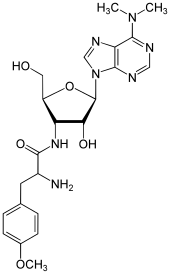
Puromycin (auch Purimycin) ist ein aus dem Bakterium Streptomyces alboniger gewonnenes Nukleosid-Antibiotikum, das die Proteinbiosynthese hemmt und gegen einige Tumore, Amöben, Trypanosomen und Würmer wirksam ist. Da es aber für den Menschen zu giftig ist, wird es nur in Experimenten der Mikrobiologie eingesetzt. Strukturell leitet es sich von Adenosin ab, von dem es sich durch zwei Methylgruppen am Purinring des Adenosins und durch eine Substitution an der Pentose unterscheidet.
Nukleosidanaloga mit Arabinose
Nukleosidanaloga mit Arabinose (Arabinosylnukleoside) werden meist als Zytostatika eingesetzt.
-
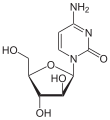 Cytarabin
Cytarabin -
 Vidarabin
Vidarabin
Anwendungsgebiete
Krebstherapie
Das älteste Anwendungsgebiet der Nukleosid-Analoga ist die Krebstherapie. Nukleosid-Analoga können als Zytostatika eingesetzt werden. Bereits 1955 wurde 6-Mercaptopurin patentiert, das zur Behandlung von Leukämien eingesetzt wurde.
| Substanz | Einsatzgebiete |
|---|---|
| Mercaptopurin | Leukämien |
| 5-Fluoruracil (5-FU) | kolorektale Karzinome, Magenkarzinom |
| Capecitabin | kolorektale Karzinome, Magenkarzinom |
| Floxuridin (FUDR), | kolorektale Karzinome |
| Trifluridin | kolorektale Karzinome (in Kombination mit dem Basenanalogon Tipiracil) |
| Cytarabin (AraC) | Leukämien, Myelodysplastisches Syndrom |
| Azaycytidin | Leukämien, Myelodysplastisches Syndrom |
| Thioguanin | Leukämien |
| Pentostatin | Haarzellleukämie |
| Fludarabin | Non-Hodgkin-Lymphome, Akute Leukämien, Chronische lymphatische Leukämie(CLL). |
| Gemcitabin | Karzinome von Pankreas, Lunge, Harnblase, Gallenwegen, Brustdrüse, Eierstock, maligne Lymphome |
| Cladribin | Haarzellleukämie |
| Decitabin | Leukämien, Myelodysplastisches Syndrom |
5-Fluoruracil (5-FU), Mercaptopurin und Thioguanin sind genau genommen keine Nukleosid-Analoga, da sie keine Pentose enthalten, sondern Basenanaloga. Alle drei Substanzen können aber nach Verabreichung als Medikament in vivo zu einem Nukleosid-Analogon umgewandelt werden.[1][2] Capecitabin ist eine Prodrug von 5-FU.
Immunsuppression
Azathioprin wird als Immunsuppressivum eingesetzt. Es wird im Körper in 6-Mercaptopurin umgewandelt.
Virostatische Therapie
Nukleosid-Analoga spielen eine entscheidende Rolle in der Therapie von Infektionen mit dem humanen Immundefizienz-Virus (HIV) und dem Hepatitis-B-Virus.
Ribavirin wirkt virostatisch nicht nur gegen das Hepatitis-C-Virus, sondern eine Reihe anderer DNA- und RNA-Viren wie beispielsweise das Respiratory-Syncytial-Virus, Influenza-Viren, Herpes-Viren, Arenaviren, Hantaviren und Adenoviren.
5-Fluoruracil wird auch topisch eingesetzt zur Behandlung von Warzen[3] und Feigwarzen (Condylomata acuminata),[4] die meist Folge einer Infektion mit humanen Papillomviren sind.
Aciclovir, Ganciclovir (oder dessen Prodrug Valganciclovir), Famciclovir, Valaciclovir, Trifluridin und Brivudin werden gegen Infektionen mit verschiedenen Herpesviren verwendet.[5][6]
Antibiotika
Bereits oben wurde Puromycin erwähnt. Eine relativ neue Entwicklung ist die Gruppe der Muramycine[7]. Das sind natürlich vorkommende antibakteriell aktive Nukleosid-Analoga, die zuerst aus Streptomyces-Bakterien isoliert wurden. Sie zeigen folgenden Aufbau: An die kettenverlängerte Ribose eines Uridin-Grundgerüsts sind verschiedene Substituenten angeknüpft. Bei einer dieser Gruppen handelt es sich um ein Peptid, das eine Harnstoff-Struktur enthält."
Thrombozytenaggregationshemmung
Das Nukleotid Adenosindiphosphat spielt auch eine Rolle bei der Aktivierung von Thrombozyten. Dabei interagiert es mit einer Familie von Rezeptoren auf Thrombozyten (P2Y1, P2Y12 und P2X1). Geeignete Nukleotid-Analoga wie Prasugrel, Clopidogrel, Ticlopidin und Ticagrelor können als Medikamente zur Hemmung der Thrombozytenfunktion (Thrombozytenaggregationshemmer) eingesetzt werden, zum Beispiel bei koronarer Herzerkrankung.
Unerwünschte Wirkungen, Wechselwirkungen und Anwendungsbeschränkungen
Wenn Nukleosid-Analoga als Medikamente eingesetzt werden, kann es zu unerwünschten Wirkungen kommen. Dabei sind wesentliche Unterschiede zwischen den als Zytostatika bzw. als Virostatika eingesetzten Nukleosid-Analoga zu beobachten. Während die Zytostatika häufig zu Übelkeit, Erbrechen und Appetitverlust sowie zu Schleimhautschäden (Mukositis) und einer Beeinträchtigung der Blutbildung führen, treten bei den Virostatika typischerweise erhöhte Leberwerte auf. Dies ist darauf zurückzuführen, dass die virostatischen Nukleosid-Analoga vor allem mit der RNA-Polymerase von Mitochondrien interagieren können[8], während die anderen Nukleosid-Analoga auch viele andere Enzyme des Nukleinsäuremetabolismus beeinträchtigen können.
Bei der gleichzeitigen Anwendungen verschiedener Nukleosid-Analoga als Medikamente drohen Arzneimittelwechselwirkungen, da das eine Nukleosid den Abbau des anderen blockieren kann. Zum Beispiel hemmt der Metabolit des Brivudins, Bromvinyluracil, irreversibel das Enzym Dihydropyrimidindehydrogenase, das für den Abbau von Pyrimidinen (wie 5-FU) zuständig ist. Allopurinol, ein Purin-Derivat und Medikament gegen zu hohe Harnsäurespiegel, verlangsamt den Abbau von Mercaptopurin und Azathioprin durch eine Hemmung der Xanthinoxidase.
Schlüsselenzyme des Abbaus verschiedener Nukleosid-Analoga weisen beim Menschen einen Polymorphismus auf, der zu einer erhöhten Toxizität führen kann, zum Beispiel bei 5-FU und Mercaptopurin. Deshalb müssen z. B. bei Menschen mit einer verminderten Aktivität der Dihydropyrimidindehydrogenase besondere Vorkehrungen getroffen werden (Dosisreduktion oder Einsatz anderer Medikamente)
Einzelnachweise
- ↑ Daniel B. Longley, D. Paul Harkin, Patrick G. Johnston: 5-Fluorouracil: mechanisms
of action and clinical strategies. In: Nature Reviews Cancer.
Band 3,
Nr. 5, Mai 2003,
S. 330–338,
doi:
 10.1038/nrc1074
( Nature).
10.1038/nrc1074
( Nature).
- ↑ Pashna N. Munshi, Martin Lubin, Joseph R. Bertino: 6-Thioguanine: A Drug With
Unrealized Potential for Cancer Therapy. In: The Oncologist.
Band 19,
Nr. 7, 2014,
S. 760–765,
doi: 10.1634/theoncologist.2014-0178,
PMID 24928612,
PMC 4077447 (freier Volltext) –
( The Oncologist).
- ↑ Sarah A Ringin: The Effectiveness of Cutaneous Wart Resolution with Current
Treatment Modalities. In: Journal of Cutaneous and Aesthetic Surgery.
Band 13,
Nr. 1, 2020,
S. 24–30,
doi: 10.4103/JCAS.JCAS_62_19,
PMID 32655247,
PMC 7335473 (freier Volltext).
- ↑ Claudio S Batista, Álvaro N Atallah, Humberto Saconato, Edina MK da Silva:
5‐FU for genital warts in non‐immunocompromised individuals. In: The Cochrane Database of Systematic
Reviews. Band 2010,
Nr. 4, 14. April 2010,
doi: 10.1002/14651858.CD006562.pub2,
PMID 20393949,
PMC 7206224 (freier Volltext).
- ↑ Celine Müller, Apothekerin, Redakteurin DAZ.online (cel):
Therapie des Herpes zoster.
28. Juni 2019.
- ↑ G. Andrei, R. Snoeck: Chapter Four - Advances in the Treatment of Varicella-Zoster
Virus Infections. In: Advances in Pharmacology (= Antiviral Agents).
Band 67. Academic Press, 1. Januar 2013,
S. 107–168,
doi: 10.1016/b978-0-12-405880-4.00004-4
( Elsevier).
- ↑ Daniel Wiegmann, Stefan Koppermann, Marius Wirth, Giuliana Niro, Kristin Leyerer:
Muraymycin nucleoside-peptide antibiotics: uridine-derived natural products as lead structures for the development of novel antibacterial agents.
In: Beilstein Journal of Organic Chemistry.
Band 12,
Nr. 1, 22. April 2016,
S. 769–795,
doi: 10.3762/bjoc.12.77,
PMID 27340469,
PMC 4902027 (freier Volltext) –
( Beilstein J Org Chem).
- ↑ Joy Y. Feng, Yili Xu, Ona Barauskas, Jason K. Perry, Shekeba Ahmadyar:
Role of Mitochondrial RNA Polymerase in the Toxicity of Nucleotide Inhibitors of Hepatitis C Virus. In:
Antimicrobial Agents and Chemotherapy. Band 60,
Nr. 2, 1. Februar 2016,
S. 806–817,
doi: 10.1128/AAC.01922-15,
PMID 26596942,
PMC 4750701 (freier Volltext) –
( ASM Journals).


© biancahoegel.de
Datum der letzten Änderung: Jena, den: 02.12. 2025