T-Lymphozyt

T-Lymphozyten oder kurz T-Zellen bilden eine Gruppe von weißen Blutzellen, die der Immunabwehr dient. T-Lymphozyten stellen gemeinsam mit den B-Lymphozyten die erworbene (adaptive) Immunantwort dar. Das T im Namen steht für den Thymus, in dem die Zellen ausreifen.
Wie alle Blutzellen werden T-Zellen im Knochenmark erzeugt. Von dort wandern sie in den Thymus, wo MHC-Rezeptoren auf ihrer Oberfläche ausgebildet werden. Durch zunächst eine positive Selektion, mit einer anschließenden negativen Selektion werden all diejenigen ausgemustert, die körpereigene MHC-Rezeptoren nicht erkennen können, oder auf körpereigene Proteine reagieren. Die restlichen, übrig gebliebenen T-Zellen können dann nur körperfremde Antigene erkennen und bekämpfen den Körper dadurch nicht selbst. Die Proteine in den selektierten Zellmembranen, auch T-Zell-Rezeptoren (TCR) genannt, können dann – ähnlich wie die von B-Lymphozyten produzierten Antikörper – körperfremde Stoffe erkennen. Im Gegensatz zu Antikörpern erkennen T-Zellen körperfremde Stoffe jedoch nur dann, wenn deren Antigene auf der Oberfläche anderer Zellen an deren MHC gebunden sind. Freie Antigene werden von T-Lymphozyten nur erkannt, wenn sie von sogenannten antigenpräsentierenden Zellen aktiv vorgezeigt werden (sog. MHC-Restriktion).
Funktion
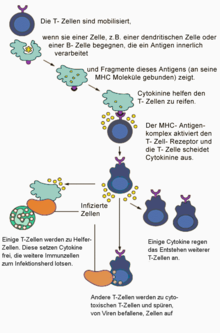
T-Zellen wandern durch den Organismus und überwachen ständig die Membranzusammensetzung der Körperzellen auf krankhafte Veränderungen. Fremdartige oder veränderte Substanzen auf der Zelloberfläche können beispielsweise durch eine Virusinfektion oder durch eine Mutation der Erbsubstanz hervorgerufen werden. Wenn eines der präsentierten MHC-I-Moleküle- oder MHC-II-Moleküle auf der Oberfläche der kranken Zelle exakt zu dem individuellen Rezeptor einer vorbeikommenden T-Zelle passt wie ein Schlüssel in das zugehörige Schloss, und wenn gleichzeitig eine Costimulanz (etwa das Oberflächenprotein B7-1) präsentiert wird, geht die T-Zelle durch Aktivierung bestimmter Gene des Zellkerns in den aktivierten Zustand über. Antigenrezeptor und Corezeptor bilden zusammen das Aktivierungssignal. Die Zelle wächst und differenziert sich zu Effektor- und Gedächtniszellen. Je nach Zellart besitzen die Effektorzellen unterschiedliche Funktionen.
„Die T-Helferzellen ermöglichen es, eine gezielte Abwehr mit maßgeschneiderten Antikörpern zu entwickeln, die T-Killerzellen töten zielgerichtet befallene Körperzellen ab, und die regulatorischen T-Zellen sorgen dafür, dass das Ganze nicht aus dem Ruder läuft. Leider sind es wohl auch bestimmte T-Zellen, die für einen besonders schweren Krankheitsverlauf von COVID-19 verantwortlich sind.“
T-Killerzellen (durch den CD8-Rezeptor gekennzeichnet) zerstören die kranke Zelle direkt; T-Helferzellen (mit CD4-Rezeptor) schlagen mit löslichen Botenstoffen (Zytokinen) Alarm und locken zusätzliche Immunzellen an. Regulatorische T-Zellen verhindern überschießende Angriffe auf intakte Körperzellen, helfen also bei der Selbsttoleranz. T-Zellen sind somit für die zellvermittelte Zytotoxizität, für die Steuerung der humoralen Immunantwort, und nicht zuletzt auch für viele allergische Reaktionen verantwortlich. Dabei hängt die Stärke der verschiedenen Reaktionen vom stimulierenden Antigen, von der Art der präsentierenden Zelle und von weiteren, zum Teil noch unbekannten Faktoren ab.
Im Thymus, einem lymphatischen Organ, werden die neuen T-Zellen für ihre unterschiedlichen Funktionen vorbereitet. Eine Einteilung kann anhand der Oberflächenantigene CD4 und CD8 erfolgen: CD4+-T-Lymphozyten werden als Helferzellen angesehen; ihr Rezeptor erkennt MHC-Klasse-II-Moleküle. CD8+-T-Lymphozyten gelten als zytotoxische T-Zellen; ihr Rezeptor erkennt Antigene, die von fast allen Körperzellen über MHC-Klasse-I-Moleküle präsentiert werden. Tatsächlich wird damit aber nur die häufigste Kombination von T-Zell-Phänotyp und -Funktion wiedergegeben; es gibt auch CD4+-zytotoxische T-Zellen und CD8+-T-Helferzellen. CD4+-T-Zellen sind vor allem im peripheren Blut und in stark durchbluteten lymphatischen Geweben wie den parafollikulären Regionen von Lymphknoten, Milz und Tonsillen zu finden. CD8+-T-Zellen kann man dagegen eher im Knochenmark und in den lymphatischen Geweben der Magen-Darm-Schleimhaut, der Atmungsorgane und der Harnwege nachweisen.
Naive (nicht aktivierte) T-Zellen bewegen sich ständig zwischen dem Blut und diesen lymphatischen Geweben. Sie besitzen zu diesem Zweck eine geringe amöboide Beweglichkeit und sind mit Zelladhäsionsmolekülen und Rezeptoren für Chemokine ausgestattet. Den Blutstrom verlassen sie mittels Diapedese durch die Wände der postkapillären Venolen. Von dort aus wandern sie durch das Gewebe und kehren mit der Lymphe über den Ductus thoracicus, der in den linken Venenwinkel mündet, wieder ins Blut zurück. Eine weitere Möglichkeit besteht darin, dass die Lymphozyten durch die Wände einer hochendothelialen Venole (HEV) in ein sekundär lymphatisches Organ einwandern.
Spezielle Funktionen der T-Lymphozyten
Immunzellen spielen eine wichtige Rolle bei der Steuerung des Knochenstoffwechsels. Sie können Substanzen freisetzen, die den Abbau der Knochenmatrix durch Osteoklasten fördern. Unter Östrogenmangel wurden im Mausmodell T-Lymphozyten zur Produktion von TNF-α und RANKL angeregt; dies könnte zur Entwicklung des Knochenmineralverlustes der Versuchstiere beigetragen haben. Thymuslose Mäuse, denen die Ovarien entfernt worden waren, erlitten trotz Hormonmangel keinen Knochenverlust. Bei thymuslosen Nacktmäusen und -ratten ist die Umsatzrate des Knochens allgemein geringer. Osteoprotegerin von aktivierten T-Lymphozyten stimuliert den osteoklastischen Abbau der Knochensubstanz und könnte an der Entstehung von Knochen- und Gelenk-Erkrankungen beteiligt sein.[2][3]
Aufbau und Unterscheidung von verwandten Zelltypen
T- und B-Lymphozyten sind kugelige Zellen von ähnlicher Größe wie rote Blutkörperchen; ihr Durchmesser beträgt beim Menschen etwa 7,5 µm. Sie können voneinander mikroskopisch oder elektronenmikroskopisch nicht unterschieden werden. Nur mittels der Immunhistochemie können Markerproteine wie das für T-Lymphozyten charakteristische CD3 und das für B-Lymphozyten spezifische CD19 dargestellt werden. Das Chromatin im runden oder leicht eingedellten, nichtgelappten Zellkern ist dicht, schollig und kräftig anfärbbar. Der Plasmasaum um den Kern ist schmal und lichtmikroskopisch kaum zu sehen. Die zahlreichen Lysosomen können als azurophile Granula sichtbar werden. Die Zellsubstanz enthält reichlich freie Ribosomen. Der Golgi-Apparat ist kleiner als bei den Retikulumzellen.
Der T-Zell-Antigenrezeptor (TCR)
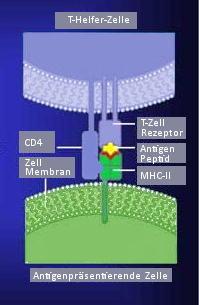
Jeder TCR auf peripheren T-Zellen ist an ein CD3-Rezeptormolekül gebunden. Der CD3-Rezeptor leitet das Aktivierungssignal in das Zellinnere. Er bindet sowohl an TCRαβ als auch TCRγδ. Das Ausmaß der Reaktion der Rezeptor- mit den Antigen-MHC-Komplexen hängt von der Konzentration beider Partner ab, d. h. ihrer Dichte auf den beteiligten Zellmembranen, und von der spezifischen Affinität des TCR. Mit der Kristallographie wurde die dreidimensionale Struktur des TCR aufgeklärt. Die antigenbindende, hypervariable V-Region ähnelt der entsprechenden V-Domäne von Antikörpern. Diese Molekülabschnitte in Form von exponierten Schleifen bestimmen die Antigenspezifität des Rezeptors und werden auch complementarity determining regions CDR genannt.
Der TCR gehört wie der Antigenrezeptor der B-Lymphozyten zur Immunglobulin-Gen-Superfamilie. Zwei von vier möglichen Proteinketten (bezeichnet mit α, β, γ, δ) sind über Disulfidbrücken verbunden. Meist ist der TCR ein αβ-Heterodimer, seltener γδ-Heterodimer. Es können also zwei Subpopulationen von T-Zellen unterschieden werden.[4] Die α-Ketten wiegen 43–49, die β-Ketten 38–44 Kilodalton, die γ-Kette 55–60 und die δ-Kette ca. 40 Kilodalton. Der Komplex aus Rezeptor und MHC ist mit 15 nm klein im Vergleich zu anderen Membranproteinen. Die Gene für die α- und δ-Kette liegen verschachtelt am gleichen Genort auf dem Chromosom 14q11-12, das γ-Ketten-Gen liegt auf Chromosom 7p15 und das β-Ketten-Gen auf Chromosom 7q32-35. Die Anordnung der Gene macht es nicht möglich, dass eine Zelle gleichzeitig Rezeptoren als γδ- und αβ-Heterodimere ausbildet.
Im Blutkreislauf und in den lymphatischen Organen gehören 95–98 % der T-Zellen der αβ-Subpopulation an. CD4+- und CD8+-T-Zellen gehören zu ihr. γδ-T-Zellen sind überwiegend (bis zu 50 %) in epithelialen Geweben wie der Haut, der Darmschleimhaut oder den Geschlechtsorganen zu finden, also an den Körperoberflächen.
Unterformen
Während in den 80er Jahren T-Zellen in die beiden Formen T-Helfer- (CD4+) und T-Suppressor-Zellen (CD8+) unterteilt wurden, kennt man inzwischen die hohe „Plastizität“ der T-Zellen, die sich in andere Subtypen wandeln oder Charakteristika mehrerer Subtypen ausbilden können, je nach vorhandenen löslichen Mediatoren, und die für den Subtyp spezifische Zytokine und Interleukine produzieren können. Subtypen sind z. B. T1, T2, T9 oder T17.[5]
T-Helferzellen
T-Zellen mit Helferfunktion sezernieren unterschiedliche Zytokine und können danach eingeteilt werden, ob diese Botenstoffe an der zellvermittelten Immunantwort beteiligt sind, oder ob die humorale Immunantwort der B-Lymphozyten stimuliert wird. So induziert die Anwesenheit von IL-12 und Interferon-γ (IFN-γ) die Differenzierung zur TH1-Zelle, während IL-4 und IL-6 eine Differenzierung zur TH2-Zelle fördern. Beispielsweise gehören unter den CD4+-Lymphozyten solche zur ersten Gruppe (Typ 1), die Interferon-γ (IFN-γ), IL-2, und TNF-α sezernieren. CD4+-Lymphozyten, die die Zytokine IL-4, IL-5, IL-6, IL-10 und IL-13 erzeugen, werden dem Typ 2 zugerechnet. Die gleiche Unterscheidung kann auch für sezernierende CD8+-T-Zellen und für solche mit einem γδ-T-Zell-Antigenrezeptor getroffen werden. Außerdem gibt es T-Helferzellen mit einem gemischten Zytokinmuster, die als Typ0-T-Zellen bezeichnet werden.
Die Unterschiede zwischen Typ1-T-Zellen und Typ2-T-Zellen wurden erstmals 1986 von Tim Mosmann beschrieben.[6]
Zytotoxische T-Zellen
Die Zytotoxischen T-Zellen (CTL) sind in der Regel durch CD8+-αβ-Heterodimere auf der Oberfläche gekennzeichnet. Sie erkennen auf MHC-I-Molekülen präsentierte Antigene, vor allem viral infizierte Zellen und Tumorzellen. CTL lösen in den defekten Zellen über deren physiologische Signalwege (Fas/FasL; Perforin/Granzyme) – den programmierten Zelltod aus.
Regulatorische T-Zellen (TReg)
Die Intensität der Immunantwort muss ständig kontrolliert werden, um einerseits die Krebszellen und Krankheitserreger zu vernichten, dabei aber Autoimmunität gegen normale Gewebe zu unterdrücken. Außerdem muss die Nachproduktion und Reifung der Leukozyten konstant gehalten werden. Ein Teil der Kontrollmechanismen wird von regulatorischen T-Zellen (veraltet Suppressor-T-Zellen) ausgeübt: über Zytokine wie IL-10 und TGF-β, durch das Abfangen von Antigenen, Wachstums- und Differenzierungsfaktoren, durch CTLA4-vermittelte Begrenzung der klonalen Expansion von B-Zellen, und durch das Abtöten von überschüssigen T-Zellen über Fas/FasL-vermittelte Signale. Die regulatorischen T-Zellen werden anhand ihrer Zytokinprofile weiter unterteilt, etwa in (CD4+-CD25+-T-reg-Zellen, TR1-Zellen, TH3-Lymphozyten und NKT-Zellen, CD8+-regulatorische Zellen).
T-Gedächtniszellen
T-Gedächtniszellen bilden eine Art „immunologisches Gedächtnis“, indem sie nach ihrer Aktivierung im Blut verbleiben. Bei einer erneuten Infektion desselben Erregers wird die ursprüngliche Aktivierung wiederhergestellt. Die Anwesenheit von Gedächtniszellen steigert die Vermehrung von antigenspezifischen T-Zellen um das 10- bis 100fache. Die Gedächtnisrolle kann sowohl durch CD4+ als auch durch CD8+-T-Gedächtniszellen ausgeübt werden.
NK-T-Zellen
Die Natürlichen Killer T-Zellen sind eine kleine Anzahl von zytotoxischen αβ-T-Zellen, die keine antigenspezifischen Rezeptoren besitzen, aber dennoch die Präsentation von MHC-I-Antigenkomplexen erkennen können. Es sind T-Zellen, man darf sie an dieser Stelle also nicht mit den Natürlichen Killerzellen des unspezifischen Immunsystems verwechseln. NK-T-Zellen sind durch das Molekül NKR-P1A, ein dem Lektin ähnliches Protein, auf ihrer Oberfläche gekennzeichnet. Weitere von NK-Zellen exprimierte Marker sind CD56, Neural cell adhesion molecule-1 (NCAM-1) und CD57. Diese Zellen produzieren auch die zytotoxischen Effektormoleküle Perforin und Granzym. Die Aufgabe der NK-T-Zellen soll in der Kontrolle von Autoimmunerkrankungen liegen.
γδ-Antigenrezeptor-positive T-Lymphozyten
γδ T-Zellen machen nur einen kleinen Prozentsatz der T-Zellen in Blut und lymphoiden Organen aus, sind aber sehr prominent in der Haut und vielen Epithel-Geweben vertreten. Ihr wesentliches Merkmal ist ein T-Zellrezeptor, der aus den γ- und δ-Untereinheiten besteht. Der γδ TCR ist deutlich weniger variantenreich als der αβ TCR, von den vielen theoretisch möglichen Kombinationen der verschiedenen Gen-Segmente werden nur einige wenige verwendet. Die Bindungspartner des γδ TCR sind noch weitgehend unbekannt, wahrscheinlich handelt es sich aber vorwiegend um körpereigene Moleküle (der αβ TCR erkennt in der Regel Antigene von Krankheitserregern).[7]
In Entwicklung und Funktion unterscheiden sich γδ T-Zellen deutlich von αβ T-Zellen. So verlassen sie den Thymus bereits in einem voraktivierten Zustand, der eine rasche Reaktion und schnelle Ausschüttung von wirksamen Substanzen ermöglicht. Die Aktivierung kann wahrscheinlich auch unabhängig vom γδ TCR durch Zytokine erfolgen. γδ T-Zellen erkennen Gewebeschäden und -veränderungen (wie etwa Krebs) und aktivieren daraufhin sowohl angeborene als auch erworbene Komponenten des Immunsystems.[8]
MAIT-Lymphozyten
MAIT-Lymphozyten (Mucosal-Associated Invariant T-Cells) sind eine Subpopulation von CD3+-Zellen mit einem semi-invarianten TCR und kommen vorzugsweise in den Schleimhäuten vor, die mit antimikrobiellen Eigenschaften ausgestattet sind.[9]
T-Lymphozyten-gebundene Erkrankungen
Angeborene Immundefekte
Ererbte Immundefekte, die sowohl die T-Zellen wie auch die B-Zellen betreffen, d. h. die zelluläre und die humorale Immunantwort schädigen, nennt man schwere kombinierte Immundefekte (SCID). Die betroffenen Kinder müssen in möglichst keimarmer Umgebung gepflegt werden und haben langfristig nur nach erfolgreicher Knochenmarktransplantation eine Überlebenschance.
Das Di-George-Syndrom verhindert die Entwicklung von Epithelgewebe im Thymus des Fetus. Daher können die T-Zellen nicht ausreifen, die zelluläre Immunreaktion ist stark vermindert.
Patienten mit Nacktes-Lymphozyten-Syndrom entwickeln Leukozyten und Thymuszellen ohne MHC-II-Moleküle und damit einen Mangel an CD4+ T-Lymphozyten.
Erworbene Immundefizienzen
Erworbene Immundefekte können durch verschiedenen Krankheiten, durch Mangelernährung, durch schädliche Effekte der Umwelt oder therapeutische Maßnahmen verursacht werden.
Infektionen
Das Humane Immundefizienz-Virus (HIV) infiziert CD4+ T-Lymphozyten, dendritische Zellen und Makrophagen, was zur Immunschwäche-Krankheit AIDS führt. Die Viren HTLV I und HTLV II können bei Menschen und Primaten T-Lymphozyten befallen und verschiedene Erkrankungen auslösen, unter anderem die Adulte T-Zell-Leukämie und die Tropische Spastische Paraparese.
Allergische Reaktionen
Von einer Überempfindlichkeitsreaktion, oder spezifischer Allergie, spricht man, wenn eine unangemessene Immunreaktion gegen körpereigenes Gewebe oder auf ein eigentlich harmloses Antigen (Staub, Pollen, Nahrungs- oder Arzneimittel) ausgelöst wird. Unter den vier Typen nach Coombs und Gell sind T-Zellen vor allem beim Typ I (Soforttyp) und Typ IV (verzögerter Typ) beteiligt. Beim Soforttyp liegt eine übersteigerte T2-Antwort gekennzeichnet, bei der verzögerten Allergie eine anhaltend übersteigerte Tätigkeit der T1-Zellen vermittelt und damit eine persistierende Entzündung.
Autoimmunerkrankungen
Autoimmunerkrankungen sind chronische Erkrankungen, die durch Immunreaktionen gegen körpereigene Antigene verursacht werden. So gibt es beim Diabetes mellitus vom Typ I die Beobachtung, dass Insulin-spezifische CD8+ T-Zellen β-Zellen des Pankreas angreifen. Auch bei der rheumatoiden Arthritis sind autoreaktive T-Zellen nachgewiesen worden.[10] Der weitverbreiteten Theorie zur Multiplen Sklerose zufolge wird auch diese Erkrankung durch aktivierte T-Zellen eingeleitet, die die Myelinscheiden der Nervenzellen zerstören.
Medikamentenwirkungen
Bestimmte Arzneimittel können erwünschte und unerwünschte Immundefizienzen hervorrufen. Nach Organtransplantationen ist die Gefahr einer Transplantabstoßung gegeben, die sowohl zelluläre wie auch humorale Immunreaktionen einbezieht. Im Vordergrund steht die T-Zell-Reaktion gegen allogene und xenogene MHC-Moleküle im fremden Gewebe. Untersuchungen zeigen drei Mechanismen: Die akute Abstoßung durch CD8+ T-Zellen, die chronische Abstoßung durch CD4+ T-Zellen, und eine Schädigung der das Transplantat versorgenden Blutgefäße. Alle drei Mechanismen können durch immunsuppressive Medikamente dauerhaft unterdrückt werden. Auch wachstumshemmende, zellabtötende Medikamente wie Zytostatika und Bestrahlungen mit ionisierender Strahlung können weiße Blutkörperchen und insbesondere T-Lymphozyten schädigen.
Onkologische Krankheitsbilder
Entartete T-Zellen sind der Ausgangspunkt einer Gruppe von Tumorerkrankungen (der malignen Lymphome), und der Akuten lymphatischen Leukämie, die oft Patienten im Kindesalter betrifft.
Das Vorkommen der T-Lymphozyten in anderen Lebewesen
In Wirbellosen (wie Einzellern, Schwämmen, Ringelwürmern und Arthropoden) finden sich weder Lymphozyten noch Lymphknoten. In Wirbeltieren kommen Lymphknoten erst bei den Vögeln und Säugetieren vor, dagegen sind die Lymphozyten schon eher im Stammbaum bei Knorpel- und Knochenfische sowie Amphibien und Reptilien vorhanden.
Forschungsgeschichte

Zu Beginn des 20. Jahrhunderts war Gegenstand der wissenschaftlichen Auseinandersetzung, ob die Immunität mancher Menschen gegen Ansteckung auf zellulären oder humoralen Vorgängen beruht. Der Zoologe Elias Metschnikow (1845–1916) beobachtete, dass sich um einen in einen Seestern gestochenen Dorn bewegliche Zellen ansammelten. Metschnikow nahm an, dass diese Zellen eingedrungene Bakterien auffressen (Phagozytosenlehre). Demgegenüber vertraten Gelehrte wie Emil Adolf von Behring (1854–1917) die Ansicht, Immunität werde durch im Blutserum gelöste Stoffe erzeugt. Bering hatte 1888 festgestellt, dass die Vermehrung von Milzbrand-Bakterien durch Serum von resistenten Ratten verhindert wird, nicht aber durch Serum von gegen Milzbrand empfindlichen Meerschweinchen. Gegen Vibrio metschnikovii wirkte nur das Serum von solchen Meerschweinchen, die zuvor mit diesem Keim infiziert gewesen waren, und deren Serum wirkte wiederum nicht gegen andere Keime. Damit konnte Behring auch Hans Buchner widerlegen, der geglaubt hatte, dass das Blutserum eine unspezifische bakterizide Aktivität habe. Gemeinsam mit Kitasato entwickelte Behring seine Lehre von der humoralen Immunität und die sogenannte „Blutserumtherapie“.

Belgische Forscher (Denys, Lecleff und Marchand), insbesondere aber Almroth Wright und S. R. Douglas konnten den scheinbaren Widerspruch beider Theorien um 1903 auflösen. Wright und Douglas fanden im Serum phagozytosefördernde Stoffe, die sie Opsonine nannten – die heutigen Antikörper – die zelluläre und humorale Vorgänge verbanden. Nach der von Linus Pauling 1940 veröffentlichten Instruktionstheorie bildeten Antigene eine Instruktion, nach der Blutzellen ein universelles Immunprotein zu einem passenden spezifischen Antikörper umformen.
Abweichend davon vertraten Niels Jerne (Klon-Selektionstheorie) und Paul Ehrlich (Seitenkettentheorie) die Ansicht, sämtliche Immunglobuline seien bereits vorgeformt und das richtige werde durch das eingeführte Antigen selektiert. Frank MacFarlane Burnet erkannte dann, dass nicht die zirkulierenden Antikörper selektiert werden, sondern einzelne immunkompetente Zellen, die dann durch Vermehrung einen spezifisch produzierenden Klon bilden (Nobelpreis 1960). Während des embryonalen Lebens entstehen durch somatische Mutationen zahllose Varianten von möglichen Antigenrezeptoren; gleichzeitig werden solche Zellen, die Rezeptoren für körpereigene Antigene tragen, wieder eliminiert.
Bis 1926 wurde die Rolle der Lymphozyten bei der Abstoßung von körperfremden Gewebe erkannt.[11][12] Gowans beschrieb 1964, dass solche Lymphozyten überall verfügbar sind, indem sie aus dem Milchbrustgang ins Blut und, über die sekundären Lymphorgane, dann wieder ins Gewebe wechseln.[13] Die besondere Bedeutung des Thymus wurde 1968 an leukämischen Mäusen entdeckt.[14] Mitte der 1960er unterschied man B- und T-Lymphozyten. Deren Zusammenspiel bei der Antikörperherstellung beschrieb Jerne 1974 (Nobelpreis 1984). 1975 unterschieden Kisielow und Mitarbeiter zytotoxische von nicht-zytotoxischen T-Zellen.[15][16] 1976 zeigten Rolf Zinkernagel und Peter Doherty, dass die T-Zelle nur aktiviert wird, wenn das auslösende Antigen am MHC präsentiert ist.[17] 1982 gelang es, einen mAb zu synthetisieren, der T-Zell-Lymphomzellen bei Mäusen erkannte.[18] Die Oberflächenstrukturen und TCRs der T-Zellen wurden genauer an T-Zell-Hybridomen und leukämischen T-Zelllinien beschrieben.[19][20] 1979 fand Kung die CD3-Proteine an der Seite des T-Zell-Antigenrezeptors;[21] deren biochemische Charakterisierung[22] folgte 1984 durch eine Forschungsgruppe von Cox Terhorst.
Der T-Zell-Rezeptor wurde 1983 in Mäusen als 45–50 kDa großes Heterodimer, mit einer α- und einer β-Kette beschrieben.[23][24] Im folgenden Jahr gelang die mRNA-Isolation auch des menschlichen TCRs, erstmals unter Zuhilfenahme der Klonierung von β-Ketten des humanen und Maus-TCRs.[25][26] Wenige Jahre später wurde ein zweiter, dem αβ-TCR ähnlicher TCR aufgefunden – der γδ-T-Zell-Antigenrezeptor.[27] Ebenfalls 1986 wurde die MHC-Restriktion des T-Zell-Antigenrezeptors erstmals beschrieben.[28] TCR-Gene können in chromosomale Mutationen einbezogen sein, die krebsfördernde Onkogene aktivieren. Mit Hilfe molekularer Proben von TCR konnten Gene identifiziert werden, die bei der Entwicklung von Leukämien und Lymphomen eine Rolle spielen.[29] 1988–1989 wurde gezeigt, dass CD8 der rezeptierende Partner für solche Antigene ist, die am MHC-I präsentiert werden. Das Gedächtnis von CD4- und CD8-Zellen wurde beschrieben.[30][31]
Literatur
- G. A. Holländer: Immunologie. Grundlagen für Klinik und Praxis. 1. Auflage. Elsevier, München 2006, ISBN 3-437-21301-6.
- M. J. Owen, J. R. Lamb: Immunerkennung. Thieme, Stuttgart 1991, ISBN 3-13-754101-8.
- I. Jahn: Geschichte der Biologie. Theorien, Methoden, Institutionen, Kurzbibliographien. 3. Auflage. Gustav Fischer, Jena 1998, ISBN 3-437-35010-2.
- A. Wollmar, T. Dingermann: Immunologie. Grundlagen und Wirkstoffe. Unter Mitarbeit von I. Zündorf. Wissenschaftliche Verlagsgesellschaft, Stuttgart 2005, ISBN 3-8047-2189-3.
- O. Bucher, H. Wartenberg: Cytologie Histologie und mikroskopische Anatomie des Menschen. 11. Auflage. Hans Huber, Bern/Stuttgart/Toronto 1992, ISBN 3-456-81803-3.
- K. Munk: Grundstudium Biologie Zoologie. Gustav Fischer, Heidelberg/Berlin 2002, ISBN 3-8274-0908-X.
Einzelnachweise
- ↑
 Die fatale Rolle der T-Zellen bei COVID-19. In: Pressemitteilung des
Berliner Institut für Gesundheitsforschung (Berlin Institute of Health at Charité, BIH), 29. Dezember 2021,:
„Originalpublikation: P. Georg, R. Astaburuaga-Garcia,…..and B. Sawitzki: Complement activation induces excessive T cell cytotoxicity in severe COVID-19“
( doi:10.1016/j.cell.2021.12.040)
Die fatale Rolle der T-Zellen bei COVID-19. In: Pressemitteilung des
Berliner Institut für Gesundheitsforschung (Berlin Institute of Health at Charité, BIH), 29. Dezember 2021,:
„Originalpublikation: P. Georg, R. Astaburuaga-Garcia,…..and B. Sawitzki: Complement activation induces excessive T cell cytotoxicity in severe COVID-19“
( doi:10.1016/j.cell.2021.12.040)
- ↑ Y. Y. Kong, H. Yoshida, I. Sarosi, H. L. Tan, E. Timms, C. Capparelli, S. Morony, A. J. Oliveira-dos-Santos, G. Van,
A. Itie, W. Khoo, A. Wakeham, C. R. Dunstan, D. L. Lacey, T. W. Mak, W. J. Boyle, J. M. Penninger: OPGL is a key regulator of
osteoclastogenesis, lymphocyte development and lymph-node organogenesis. In: Nature.
Band 397, Nr. 6717, 28. Januar 1999, S. 315–323,
PMID 9950424 (englisch)
- ↑ S. Cenci, M. N. Weitzmann, C. Roggia, N. Namba, D. Novack, J. Woodring, R. Pacifici:
Estrogen deficiency induces bone loss by enhancing T-cell production of TNF-alpha. In:
Journal of Clinical Investigation.
Band 106,
Nr. 10, November 2000,
S. 1229–1237,
PMID 11086024 (englisch).
- ↑ M. Girardi: Immunosurveillance and immunoregulation by
γδ T cells. In: Journal of Investigative Dermatology.
Nr. 126, 2006,
S. 25–31,
PMID 16417214 (englisch).
- ↑ Stefanie Sarantopoulos: Allogenic stem-cell Transplantation – a T-cell
balancing ACT. In: New England Journal of Medicine.
Band 378,
Nr. 5, 1. Februar 2018,
S. 480–482,
doi: 10.1056/NEJMcibr1713238 (englisch).
- ↑ Tim Mosmann, H. Cherwinski, M. W. Bond, M. A. Giedlin, R. L. Coffman: Two
types of murine helper T cell clone. I. Definition according to profiles of lymphokine activities and secreted proteins. In:
Journal of Immunology. Band 136,
Nr. 7, 1986,
S. 2348–2357 (englisch,
Abstract).
- ↑ M. Bonneville, R. L. O’Brien, W. K. Born: Gammadelta T cell effector
functions: a blend of innate programming and acquired plasticity. In: Nature Reviews Immunology.
Band 10, 2010,
S. 467–478,
PMID 20539306 (englisch).
- ↑ A. C. Hayday: Gammadelta T cells and the lymphoid stress-surveillance
response. In: Immunity.
Band 31, 2009,
S. 184–196,
PMID 19699170 (englisch).
- ↑ Lionel Le Bourhis, Emmanuel Martin, Isabelle Péguillet, Amélie Guihot, Nathalie Froux, Maxime Coré,
Eva Lévy, Mathilde Dusseaux, Vanina Meyssonnier, Virginie Premel, Charlotte Ngo, Béatrice Riteau, Livine Duban, Delphine Robert, Shouxiong Huang, Martin Rottman, Claire Soudais,
Olivier Lantz: Antimicrobial activity of mucosal-associated invariant T cells. In:
Nature Immunology. Band 11,
Nr. 8, August 2010,
S. 701–708,
doi: 10.1038/ni.1890,
PMID 20581831 (englisch).
- ↑ György Nagy, Joanna M. Clark, Edit Buzas, Claire Gorman, Maria Pasztoi, Agnes Koncz,
András Falus, Andrew P. Cope: Nitric oxide production of T lymphocytes is increased in rheumatoid arthritis. In:
Immunology Letters. Band 118,
Nr. 1, 2008,
S. 55–58,
doi: 10.1016/j.imlet.2008.02.009 (englisch).
- ↑ J. B. Murphy: Studies in tissue specifity. II. The ultimate fate of mammalian tissue implanted in chick embryo. In: Journal of Experimental Medicine. Nr. 19, 1914, S. 181–186 (englisch).
- ↑ J. B. Murphy: Factors of resistance to heteroplastic tissue-grafting. Studies in tissue specifity III. In: Journal of Experimental Medicine. Nr. 19, 1914, S. 513–522 (englisch).
- ↑ J. L. Gowans, E. J. Knight: The route of re-circulation of lymphocytes
in the rat. In: Proceedings of the Royal Society of London B. Biol. Sci.
Nr. 159, 1964,
S. 257–282,
PMID 14114163 (englisch).
- ↑ J. F. Miller: Immunological function of the thymus. In: The Lancet. Nr. 2, 1968, S. 748–749 (englisch).
- ↑ P. Kisielow, J. A. Hisrst, H. Shiku, P. C. Beverley, M. K. Hoffman, E. A. Boyse, H. F. Ottgen:
Ly antigens as markers for functionally distinct subpopulations of thymus-derived lymphocytes of the mouse. In:
Nature. Nr. 253, 1975,
S. 219–220,
PMID 234178.
- ↑ H. Shiku, P. Kisielow, M. A. Bean, T. Takahashi, E. A. Boyse, H. F. Ottgen, L. J. Old:
Expression of T-cell differentiation antigens on effector cellsin cell-mediated cytotoxicityin vitro. In:
Journal of Experimental Medicine.
Nr. 141, 1975,
S. 227–241,
PMID 1078839 (englisch).
- ↑ R. M. Zinkernagel, P. C. Doherty: Restriction of in vitro T-cell
mediated cytotoxicity in lymphocytic choriomeningitis within a syngeneic or semiallogeneic system. In: Nature.
Nr. 248, 1974,
S. 701–702,
PMID 4133807 (englisch).
- ↑ J. P. Allison, B. W. McIntyre, D. Bloch: Tumor specific antigen of
murine T-lymphoma defined with monoclonal antibody. In: Journal of Immunology.
Nr. 129, 1982,
S. 2293–2300,
PMID 15661866 (englisch).
- ↑ L. E. Samelson, R. N. Germain, R. H. Schwatz: Monoclonal antibodies
against the antigen receptor on a cloned T-cell hybrid. In:
Proceedings of the National Academy of Sciences of the United States of America.
Nr. 80, 1983,
S. 6972–6976,
PMID 6316339 (englisch).
- ↑ R. D. Bigler, D. E. Fischer, C. Y. Wang, E. A. Kan, E. A. Rinnooy Kan, H. G. Kunkel:
Idiotype-like molecules on cells of human T cell leukemia. In:
Journal of Experimental Medicine.
Nr. 158, 1983,
S. 1000–1005,
PMID 6604124 (englisch).
- ↑ P. Kung, G. Goldstein, E. Reinherz, S. F. Schlossman: Monoclonal antibodies defining distinctive human T cell surface antigens. In: Science. Band 206, 1979, S. 347–349 (englisch).
- ↑ H. C. Oettgen, J. Kappler, W. J. M. Tax, C. Terhorst: Characterisation
of the two heavy chains of the T3 complex on the surface of human T lymphocytes. In: The Journal of Biological Chemistry.
Band 259,
Nr. 19, 10. Oktober 1984,
S. 12039–12048,
PMID 6090452 (englisch).
- ↑ O. Acuto, R. E. Hussey, K. A. Fitzgerald, J. P. Protentis, S. C. Meuer, S. F. Schlossman, E. L. Reinherz:
The human T cell receptor: appearance in ontogeny and biochemical relationship of alpha and beta subunits on IL-2 dependent clones and
T cell tumors. In: Cell.
Nr. 34, 1983,
S. 717–726,
PMID 6605197 (englisch).
- ↑ J. Kappler, R. Kubo, K. Haskins, J. White, P. Marrack: The mouse T cell
receptor: comparison of MHC-restricted receptors on two cell hybridomas. In: Cell.
Nr. 34, 1983,
S. 727–737,
PMID 6605198 (englisch).
- ↑ Y. Yanagi, Y. Yoshikai, S. P. Clark, I. Aleksander, T. W. Mak: A human
T cell-specific cDNA clone ancodes a protein having extensive homology to immunoglobulin chains. In: Nature.
Nr. 308, 1984,
S. 145–149,
PMID 6202421 (englisch).
- ↑ S. M. Hedrick, D. I. Cohen, E. A. Nielsen, M. M. Davis: Isolation of
cDNA clones encoding T cell specific membrane-associated proteins. In: Nature.
Nr. 308, 1984,
S. 149–153,
PMID 16116160 (englisch).
- ↑ M. B. Brenner, J. McLean, D. P. Dialynas, J. L. Strominger, J. A. Smith, F. L. Owen, J. G. Seidman et al.:
Identification of a putative second T cell receptor. In: Nature.
Nr. 322, 1986,
S. 145–149,
PMID 3755221 (englisch).
- ↑ Z. Dembic, W. Haas, S. Weiss, J. McCubrey, H. Kiefer, H. von Boehmer, M. Steinmetz:
Transfer of specifity by murine alpha and beta T cell receptor genes. In: Nature.
Nr. 320, 1986,
S. 232–238,
PMID 2421164 (englisch).
- ↑ W. H. Lewis, E. E. Michalopoulos, D. L. Williams, M. D. Minden, T. W. Mak:
Breakpoints in the human T cell antigenreceptor alpha chainlocus in two T-cell leukemia patients with chromosomal translocation. In:
Nature. Nr. 317, 1985,
S. 544–546,
PMID 3876514 (englisch).
- ↑ A. M. Norment, R. D. Salter, P. Parham, V. H. Engelhard, D. R. Littman:
Cell-cell adhesion mediated by CD8 and MHC-I class I molecules. In: Nature.
Nr. 336, 1988,
S. 79–81,
PMID 3263576 (englisch).
- ↑ D. Maspoust, V. Vezys, E. J. Wherry, R. Ahmed: A brief history of CD8 T
cells. In: European Journal of Immunology.
Nr. 37, 2007,
S. 103–110,
PMID 17972353 (englisch).


© biancahoegel.de
Datum der letzten Änderung: Jena, den: 22.05. 2026