Hochleistungsflüssigkeitschromatographie
Hochleistungsflüssigkeitschromatographie (englisch high performance liquid chromatography, HPLC) – in den Anfangszeiten dieser Technik auch Hochdruckflüssigchromatographie (englisch high pressure liquid chromatography) genannt – ist eine analytische Methode in der Chemie. Die HPLC ist ein Flüssigchromatographie-Verfahren, mit dem man nicht nur Substanzen trennt, sondern diese auch über Standards identifizieren und quantifizieren (die genaue Konzentration bestimmen) kann. Im Unterschied zur Gaschromatographie, die eine sehr gute Trennmethode für verdampfbare Stoffe ist, können mittels HPLC auch nicht-flüchtige Substanzen analysiert werden. Die HPLC kann auch präparativ genutzt werden.
HPLC wurde in den 1960er Jahren entwickelt. Zu den Pionieren zählen Joseph Jack Kirkland, Josef Franz Karl Huber, Csaba Horváth und John Calvin Giddings.[1]
Funktionsweise
Es handelt sich um ein chromatographisches Trennverfahren, bei dem die zu untersuchende Substanz zusammen mit einem Laufmittel, der mobilen Phase (auch „Elutionsmittel“ oder „Eluent“ genannt) durch eine sogenannte Trennsäule gepumpt wird, welche die stationäre Phase enthält. Eine Trennsäule in einem HPLC-Gerät ist zwischen 18 und 300 mm lang und hat zumeist einen Innendurchmesser von 2 bis 4,6 mm im Falle von analytischen HPLC-Systemen. Das Trennvermögen einer HPLC ist etwa 100-mal größer als in der Säulenchromatographie. Häufig wird aus wirtschaftlichen Gründen eine sogenannte Vorsäule oder ein Säulenfilter vorgeschaltet. Dabei handelt es sich um eine kurze Säule oder eine Filterscheibe aus gleichem Material wie die Trennsäule, um Verunreinigungen von der Hauptsäule abzuhalten. Die HPLC findet auch Verwendung für die Reinigung von Substanzen als (semi-)präparative HPLC. Die Innendurchmesser können erheblich größer sein, da bis hin zum Produktionsmaßstab eine Aufreinigung durchgeführt werden kann. Präparative Säulen für den Labormaßstab haben einen Durchmesser von 10 oder 25 mm.
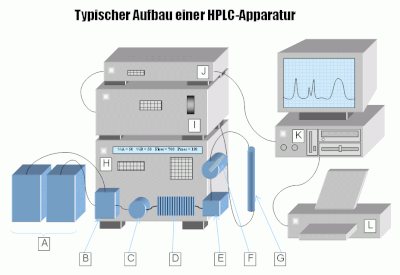
A = Eluentenreservoirs
B = Elektromagnetische Mischventile mit Doppelhubkolbenpumpe
C = 6-Wege-Ventil
D = Druckkompensationsschleife, um Pumpimpulse der Pumpe zu egalisieren
E = Mischkammer
F = Manuelles Einspritzventil
G = Trennsäule
H = HPLC-Einheit
I = Detektor-Einheit (z. B. UV-Spektrometer)
J = Computer-Interface
K = PC
L = Drucker zur Ausgabe der Ergebnisse
Der auf die wesentlichen Elemente reduzierte Aufbau einer typischen HPLC-Apparatur kann der nebenstehenden Abbildung entnommen werden. Man unterscheidet nach dem Trennprinzip in Normalphasen- (engl. normal phase, NP), Umkehrphasen- (engl. reverse phase, RP), Ionenaustausch- (IEC) und Größenausschlusschromatografie (engl. size exclusion chromatography, SEC) sowie Enantiomerentrennung (chiral chromatography).
Wechselwirkt ein Bestandteil der zu untersuchenden Substanz stark mit der stationären Phase, verbleibt er relativ lange in der Säule. Wechselwirkt er hingegen schwach mit der stationären Phase, verlässt er die Säule früher. Je nach Stärke dieser Wechselwirkungen erscheinen die Bestandteile der Substanz zu verschiedenen Zeiten (den Retentionszeiten) am Ende der Trennsäule, wo sie dann mit einem geeigneten Detektor nachgewiesen werden können. Für die Umkehrphase ist die Retentionszeit einer Substanz abhängig von der Verweildauer in der stationären Phase (Lösungsmittelfilm um die Alkylketten des modifizierten Kieselgels). Der geschwindigkeitsbestimmende Schritt ist die „Zurücklösung“ (Desorption) in die mobile Phase.
Bei der NP-HPLC wird eine polare stationäre Phase (z. B. Silicagel/Kieselgel) genutzt. Die Stärke der Elutionskraft der mobilen Phase ist im Allgemeinen abhängig von der Polarität. Die verschiedenen Lösungsmittel sind nach steigender Polarität in der elutropen Reihe angeordnet. Je polarer eine mobile Phase ist, desto schneller wird eine Substanz eluiert. Polare Moleküle werden auf der Säule länger adsorbiert/retardiert (zurückgehalten) als unpolare Moleküle und verlassen deshalb die Säule später. Die Normalphasen haben den Nachteil, dass man meist nur mit organischen Lösungsmitteln und nicht mit wässrigen Eluenten arbeiten kann. Einen Ausweg aus diesem Problem bietet die hydrophile Interaktionschromatographie (HILIC). In der HILIC werden analog zur NP-Selektivität polare stationäre Phasen verwendet, die aber mit wässrigen Puffersystemen als Eluent funktionieren. Im Gegensatz zur RP-Chromatographie ist jedoch in der HILIC Wasser das stärkste Elutionsmittel.
Die RP-HPLC ist in der Praxis die gängigste Methode. Etwa 70 % aller analytischen HPLC-Trennungen sind RP-Trennungen. Hier wird eine unpolare stationäre Phase verwendet, und die Elutionskraft der mobilen Phase sinkt mit steigender Polarität. Die stationäre Phase wird hergestellt, indem man Silane, welche mit langkettigen Kohlenwasserstoffen substituiert wurden, mit Silicagel reagieren lässt. Dabei wird die polare Oberfläche der Silicagel-Partikel mit einer unpolaren Schicht aus Alkanen überzogen, also die Polarität verringert. Als mobile Phase werden meist Mischungen aus Wasser oder Puffer und Acetonitril oder Methanol eingesetzt. Bei isokratischen Trennungen bleibt die Zusammensetzung der mobilen Phase während der gesamten Zeit gleich. Bei Gradiententrennungen wird die Polarität des Fließmittelgemisches während der Analyse verändert.
Besondere Anwendung findet die RP-HPLC bei der Auftrennung von polaren Analyten, die auf Normalphasen zu hohe Retentionszeiten aufweisen würden. Dafür wird meist eine C18-Säule (also ein Octadecylsilan als Derivatisierungsreagenz für das Silicagel, daher auch die häufige Bezeichnung ODS-Säule[2]) eingesetzt. Die Detektion erfolgt zumeist mittels UV- oder Fluoreszenzdetektor. Immer häufiger kommen Kombinationen mit einfachen Massenspektrometern (MS) oder Tandem-Massenspektrometern zum Einsatz.
Praktische Durchführung und Anwendungen
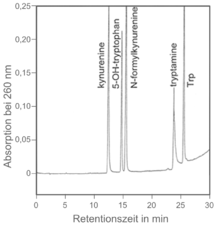
Eine chemische Verbindung kann man mittels HPLC nur bedingt identifizieren, indem man die Retentionszeit der unbekannten Substanz mit der eines Standards (einer bekannten Substanz) vergleicht (externe Standardisierung). Ist die Retentionszeit gleich, kann man der Probe mit der unbekannten Substanz auch etwas Standard zusetzen und untersuchen, ob beim Chromatogramm nach wie vor nur eine Spitze (engl. peak) sichtbar ist, ob ein „Doppel-Peak“ entstanden ist oder ob am Chromatogramm zwei getrennte „Peaks“ mit sehr ähnlicher Retentionszeit sichtbar werden (interne Standardisierung). Wenn nach Zusatz von Standard zur Probe nur eine Spitze sichtbar ist, kann man noch nicht davon ausgehen, dass die chemische Verbindung in der Probe und im Standard identisch ist. Ein weiterer Parameter wäre der Abgleich des UV-Spektrums bei Verwendung eines Diodenarray-Detektors oder der Massenspur bei einem gekoppelten Massenspektrometer. Allerdings bieten Retentionszeit, UV-Spektrum und MS-Spektrum bei Isomeren oft nur unzureichende Identifikationsmerkmale. Hier kann diese Technik in der Praxis die effiziente Identifizierung nur erleichtern, indem sie sehr gut andere Möglichkeiten ausschließt.
Alternativ führt man dieses Experiment unter zwei unterschiedlichen Trennbedingungen (z. B. HPLC-Trennungen mit zwei unterschiedlichen Säulen) durch. So kann man damit unbekannte chemische Verbindungen mit einer gewissen Sicherheit identifizieren.
Will man die Konzentration einer chemischen Substanz bestimmen (z. B. von Vitamin E in einem pflanzlichen Öl), so kann man dies tun, indem man Standards dieser chemischen Substanz mit bekannten Konzentrationen herstellt und die Peak-Fläche der Standards mit den Peak-Flächen der Substanz in den Proben vergleicht. Wie bei jedem analytischen Verfahren ist darauf zu achten, dass bei einer vorangehenden Probenaufarbeitung die Wiederfindungsrate mit in die Berechnung der Konzentration einbezogen wird.
Chromatographische Trennungen werden nicht ausschließlich zu analytischen Zwecken eingesetzt, sondern auch für präparative Zwecke im Labor und in der chemischen Industrie, um ein Produkt (z. B. Proteine) zu reinigen oder sehr ähnliche Substanzen (z. B. Enantiomere) voneinander zu trennen. Hierbei kommen Säulen mit bis zu einem Meter Durchmesser zum Einsatz.
Methodenentwicklung
Die Trennung der Substanzen ist von vielen Parametern abhängig, darunter
- Art der Substanzen,
- Art und Maße der Trennsäule,
- Zusammensetzung der mobilen Phase,
- Temperatur,
- pH-Wert
- Durchflussgeschwindigkeit der mobilen Phase
Zum Erreichen einer vollständigen und reproduzierbaren Trennung muss meist für jedes komplexere Stoffgemisch, gerade bei der Gradientenmethode, eine eigene Methode entwickelt werden. Schon geringe Abweichungen von einer Methode können eine Änderung der Selektivität bedeuten. Ziel einer Methodenentwicklung ist ein Chromatogramm, bei dem alle Spitzen vollständig getrennt sind, jedoch in minimalem Abstand voneinander auftauchen, um die Dauer eines Trennvorgangs möglichst gering zu halten. Um die Parameter nicht zeitaufwändig und kostspielig durch Versuch und Irrtum (engl. trial and error) bestimmen zu müssen, bedienen sich chemische, pharmazeutische und die Nahrungsmittelindustrie oft einer Simulationssoftware, die, auf experimentellen Daten oder Molekülstrukturen der Proben beruhend, passende Methoden prognostizieren kann.
Aktuelle Entwicklungen: UHPLC
In den letzten Jahren ging der Trend zu immer höherem Probendurchsatz mit immer kleineren Probenvolumina. Als neutrale Bezeichnung für HPLC mit stark gesteigerter Leistung ist die Abkürzung UHPLC (kurz für engl. Ultra High Performance Liquid Chromatography) geeignet. Diese Bezeichnung ist analog zu „High Frequency“ (HF) und „Ultra High Frequency“ (UHF) oder „High Temperature“ (HT) und „Ultra High Temperature“ (UHT) zu verstehen. Verschiedene Hersteller von HPLC-Anlagen haben dagegen unterschiedliche Abkürzungen und Begriffe geprägt:
- RRLC: Rapid Resolution Liquid Chromatography
- RSLC: Rapid Separation Liquid Chromatography
- UFLC: Ultra Fast Liquid Chromatography
- UPLC: Ultra Performance Liquid Chromatography
- ULDC: Ultra Low Dispersion Chromatography
Allen Techniken gemeinsam ist, dass hierbei Partikel mit einem Durchmesser von 2,2 bis 1,7 μm als Säulenmaterial genutzt werden. Dadurch können Geschwindigkeit und Effizienz einer chromatographischen Trennung deutlich verbessert werden. Eine typische UHPLC-Analytik, inkl. Gradientenelution und anschließender Equilibrierung dauert 5 bis 10 Minuten (Faustregel 1/10 der HPLC), wobei Probenvolumina von 0,5 bis 2 µl verwendet werden. Es gibt aber auch etablierte Methoden, mit einer Analysezeit von weniger als einer Minute. Für die Durchführung einer Trennung mit einem solchen Säulenmaterial ist ein deutlich höherer Arbeitsdruck von bis zu 1000 Bar[3] erforderlich, der von klassischen HPLC-Anlagen nicht erreicht werden kann. Sowohl die Säulen als auch die anderen Bauteile wie Injektionsgeber, Pumpen, Ventile und Verbindungselemente müssen unter diesen Bedingungen zuverlässig arbeiten.[4]
Bei diesen Verfahren wird die Analysezeit durch optimierte Anlagenbauteile verkürzt (z. B. schnelle, verschleppungsarme Probengeber, kleinere Kapillaren, empfindliche Detektoren, geringe Gradientenverzögerung).
Die erste UHPLC-Anlage wurde unter dem Namen UPLC™ von der Firma Waters Corporation 2004 auf den Markt gebracht. Inzwischen ist die UHPLC eine Standardmethode und im Begriff die klassische HPLC in weiten Teilen abzulösen. Dazu trägt im Wesentlichen die immer größer werdende Verbreitung sowie der einfache Methodentransfer bei.
Simulated moving bed (SMB)–Technologie
Ein weiteres hocheffektives Trenn- und Reinigungsverfahren, speziell in der Naturstoff- oder Arzneistoffanalytik. Es wird das Prinzip der kontinuierlichen Gegenstromverteilung angewandt.[5][6]
Detektoren
- UV/VIS-Detektor
- Diodenarraydetektor (DAD)
- Multiwellenlängendetektor (MWD)
- Lichtstreudetektor
- Fluoreszenzdetektor
- Brechungsindexdetektor
- Massenspektrometer
- Leitfähigkeitsdetektor
- Elektrochemischer Detektor: Amperometrie, Coulometrie
- Radioaktivitäts-Detektor
- Stickstoffselektiver Detektor
Literatur
- Gaby Aced, Hermann J. Möckel: Liquidchromatographie – Apparative, theoretische und methodische Grundlagen der HPLC, VCH, Weinheim 1991, ISBN 3-527-28195-9
- Heinz Engelhardt (Hrsg.): Practice of High Performance Liquid Chromatography. Applications, Equipment and Quantitative Analysis. Springer, Berlin u. a. 1986, ISBN 3-540-12589-2.
- Hans Henke: Flüssig-Chromatographie. Analytische und präparative Trennungen. Vogel-Buchverlag, Würzburg 1999, ISBN 3-8023-1757-2.
- Henk Lingeman, Willy J. M. Underberg (Hrsg.): Detection-Oriented Derivatization Techniques in Liquid Chromatography (= Chromatographic Science. Bd. 48). Marcel Dekker Inc., New York NY u. a. 1990, ISBN 0-8247-8287-9.
- Reinhard Matissek, Reiner Wittkowski (Hrsg.): High Performance Liquid Chromatography in Food Control and Research. Behr’s Verlag, Hamburg 1992, ISBN 3-86022-029-2.
- Veronika R. Meyer: Fallstricke und Fehlerquellen der HPLC in Bildern. Hüthig GmbH, Heidelberg 1995, ISBN 3-7785-2417-8.
- Lloyd R. Snyder, Joseph J. Kirkland, John W. Dolan: Introduction to Modern Liquid Chromatography. 3rd Edition. John Wiley & Sons, Hoboken NJ 2010, ISBN 978-0-470-16754-0.
Einzelnachweise
- ↑ Derek Lowe, Das Chemiebuch, Librero 2017, S. 426
- ↑ HPLC für Neueinsteiger (PDF, 296 kB)
- ↑ Lucie Nováková, Ludmila Matysová, Petr Solich: Advantages of application of UPLC in pharmaceutical analysis. In: Talanta. Band 68, Nr. 3, S. 908–918, doi:10.1016/j.talanta.2005.06.035.
- ↑ Ultra Low Dispersion Chromatography
- ↑ Faria RP, Rodrigues AE: Instrumental aspects of Simulated Moving Bed chromatography., J Chromatogr A. 2015 Nov 20;1421:82-102, PMID 26341597
- ↑ Caes BR, Van Oosbree TR, Lu F, Ralph J, Maravelias CT, Raines RT: Simulated moving bed chromatography: separation and recovery of sugars and ionic liquid from biomass hydrolysates. T. ChemSusChem. 2013 Nov;6(11):2083-9, PMID 23939991


© biancahoegel.de
Datum der letzten Änderung: Jena, den: 09.06. 2025